Cluster Analysis on the Single Cell Level
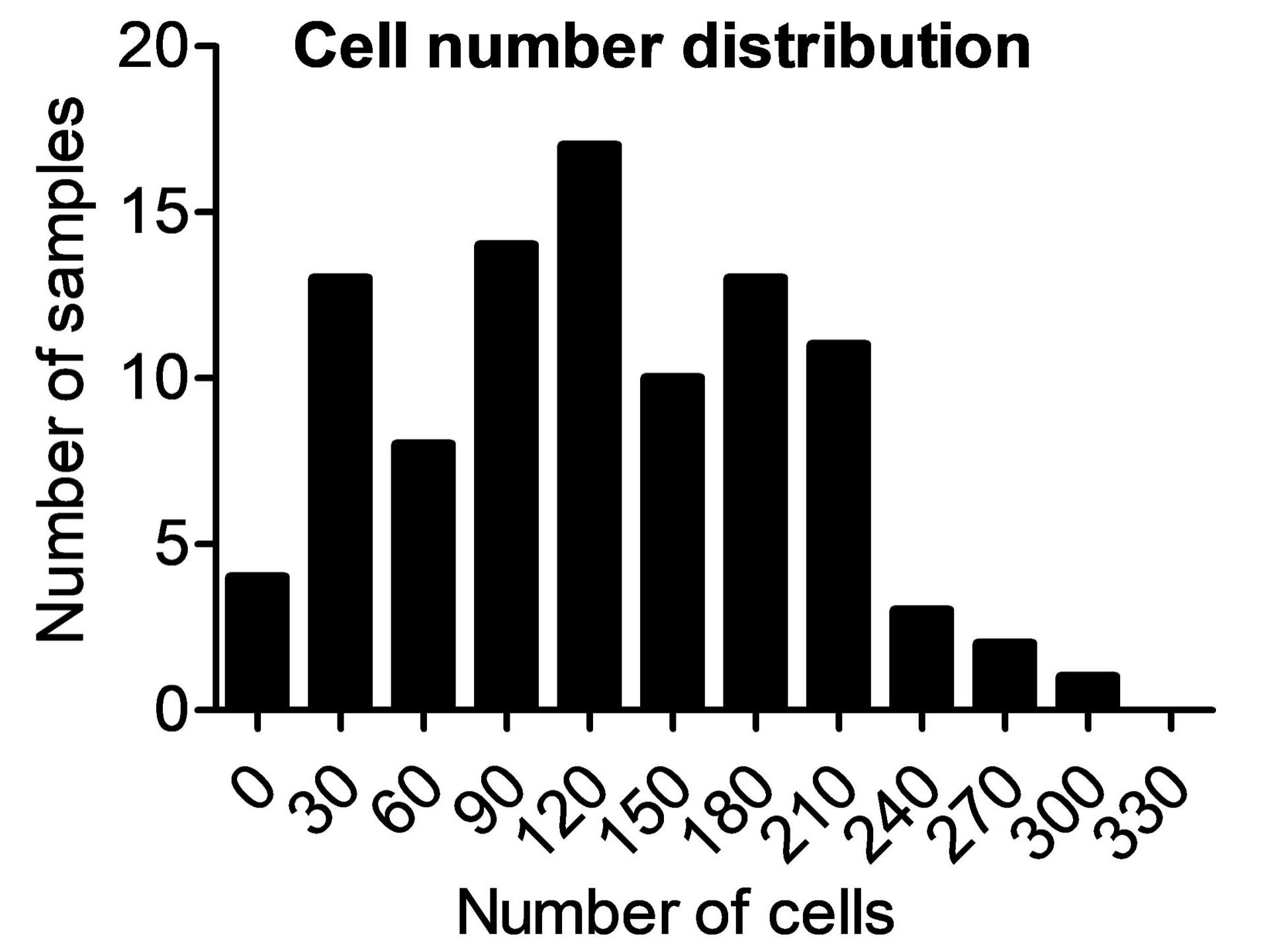
Normally at this stage of the study, we would examine single quantitative features and plot them, thereby describing observations and formulating biological conclusions. However, having obtained multidimensional quantitative output from this data set allowed us to apply more advanced data analysis tools. With such approaches, all features are examined together in an objective fashion to find systematic deviations, if any exist. Cluster analysis is a method that categorizes cells or samples based on the entirety of extracted features and sorts them into groups exhibiting the highest similarity.
Features in this data set vary over several orders of magnitude, with some displaying very large values (e.g., the pixel sum intensities of the nucleus) while other values are rather small (e.g., actin area fraction). For the cluster algorithm, the data needs to be normalized. This was achieved by dividing every value by the average value, then by the standard deviation of the specific feature. Here, we additionally log-transformed the data set.
To clarify the large number of data points generated, the results are then typically plotted as a heatmap. Figure 9A shows the heatmap for every single cell grouped by the sample they belong to (= siRNA treatments; notice the SampleID color code). The feature pattern shows that the experimental features vary considerably between samples but are consistent within a sample. Figure 9B shows the same data clustered for feature similarity. It becomes apparent that cells from many different samples will cluster into groups of similar phenotypes. Examining the features within these clusters shows that different feature combinations can be observed. There is, for example, a cluster that shows high abundance of actin and microtubules, and yet another cluster shows high abundance of focal adhesions.
Of note in Figure 9B, not only cells are clustered by similarity in Figure 9B, but also the underlying measurement features. Features that occur in the same cluster show similar responses for all samples. Examining feature clusters allows identification of common regulation patterns. For example, when we see a feature cluster for nucleus intensity and area, it means, of course, that they are most likely commonly regulated. The features “Single Focal adhesion intensity” and “Focal adhesion numbers per cell” do not occur in the same cluster and thus are likely not commonly regulated, which might come as a surprise. Many other observations can be revealed with further analysis of the data set, but these will not be discussed here for the sake of brevity. However, using the criteria presented in this example, we hope to illustrate that such correlative analysis can be information-rich and revealing.





































 is shown. Sample IDs are shown with every image.")
 is shown. Sample IDs are shown with every image.")
 is shown. Sample IDs are shown with every image.")
 is shown. Sample IDs are shown with every image.")
 is shown. Sample IDs are shown with every image.")
 is shown. Sample IDs are shown with every image.")
 is shown. Sample IDs are shown with every image.")
 is shown. Sample IDs are shown with every image.")
. Sample IDs are shown with every image.")
. Sample IDs are shown with every image.")
. Sample IDs are shown with every image.")
. Sample IDs are shown with every image.")


.")
.")









